





Mukopolysaccharidose
Mukopolysaccharidosen stellen eine große Gruppe der erblichen Monogenkrankheiten dar, die infolge eines Defekts entstehen. Der Defekt liegt in einem Lysosom vor und betrifft den mehrstufigen Prozess der Glykosaminoglykan-Hydrolyse.
 des Verstoßes in lisossomach des mehrstufigen Prozesses der zymischen Katalyse glikosaminoglikanow entstehen (die Eiderenten) vor.
des Verstoßes in lisossomach des mehrstufigen Prozesses der zymischen Katalyse glikosaminoglikanow entstehen (die Eiderenten) vor.
Glykosaminoglykane sind komplexe Heterozuckerketten, die aus Resten sulfatierten Hexosamins sowie Glukuronsäure bestehen. Zu den Glykosaminoglykanen zählen Dermatansulfat, Keratansulfat, Heparansulfat, Chondroitin-6-Sulfat sowie Chondroitin-4-Sulfat.
Die Mukopolysaccharidose wurde erstmals 1917 von Gurler beschrieben. Heute sind elf genetische Formen bekannt: fünf entstehen durch Defekte der Sulfatasen, vier durch Defekte der Glycosidasen und ein Typ durch einen Transferasemangel. Bei dieser Erkrankung kommt es zur Ansammlung von Substanzen im Organismus, was beim Menschen zu verschiedenen Symptomen und Pathologien führt.
Die Erkrankung wird autosomal-rezessiv vererbt.
Bei der Mukopolysaccharidose kommt es durch Defekte an Sulfatasen, Glycosidasen oder Transferasen zur Ansammlung von Substanzen im Organismus. Dies führt beim Menschen zu verschiedenen Symptomen und Pathologien.
Bei dieser Erkrankung kommt es zu einer Störung des lysosomalen Enzymsystems, das am Abbau von Glykosaminoglykanen beteiligt ist. Das Enzymdefizit führt in Organen und Geweben zur Ansammlung von Glykosaminoglykanen, wodurch die Mukopolysaccharidose als Speicherkrankheit klassifiziert wird.
Die Akkumulation der Glykosaminoglykane beeinträchtigt den funktionellen Zustand verschiedener Systeme und Organe. Da diese Substanzen das Bindegewebe infiltrieren, stellt eine systemische Skelettinfektion sowie eine Hemmung der Körperentwicklung eines der Hauptsymptome dar – insbesondere bei Typ I, IV und VI.
Die Symptome der Mukopolysaccharidose
Bei dieser Erkrankung fällt eine Wachstumsverzögerung auf, die gegen Ende des ersten Lebensjahres einsetzt. Zu den groben Merkmalen gehören: große Zunge, hängende Stirnpartie, Zahn- und Ohrenfehlstellungen sowie Hypertelorismus. Der Brustkorb ist deformiert; Kyphose der Wirbelsäule (Lenden- und Brustbereich) ist deutlich ausgeprägt. Charakteristisch sind Hepatosplenomegalie, Inguinal- und Nabelhernien sowie eine Einschränkung der Gelenkbeweglichkeit.
Bei der radiologischen Untersuchung lassen sich 'fischgrätenartige' Wirbelkörper sowie eine frühzeitige Ossifikation des Okzipito-parietalen Nahtbereichs erkennen. Die Bildung der Ossifikationskerne ist dabei nicht gestört.
Zu den neurologischen Symptomen der Mukopolysaccharidose gehören allgemeine Bewegungsstörungen, die diffuse Muskelhypotonie umfassen können. Für verschiedene Typen der Mukopolysaccharidose sind zudem eine Abschwächung des Gehörs und eine Senkung des Intellekts charakteristisch.
 Muskelhypotonie. Für verschiedene Typen mukopolissacharidosa auch ist die Abschwächung des Gehörs und die Senkung des Intellekts charakteristisch.
Muskelhypotonie. Für verschiedene Typen mukopolissacharidosa auch ist die Abschwächung des Gehörs und die Senkung des Intellekts charakteristisch.
Die Typen der Mukopolysaccharidosen
Nach dem Grad der Ausprägung psychischer Symptome und Knochenveränderungen sowie nach der Geschwindigkeit des Fortschreitens der Austauschstörungen sind sieben Formen dieser Erkrankungen bekannt:
- Typ I, auch als Gurlers Syndrom bezeichnet: Die Erkrankung entwickelt sich rasch. Im Urin der Patienten findet sich ein hoher Gehalt an Chondroitinsulfat und Heparansulfat. Die Vererbung erfolgt autosomal-rezessiv.
- Typ II oder das Syndrom von Hunter: Bei dieser Erkrankung treten Pigmentretinitis und Schwerhörigkeit auf. Der Krankheitsverlauf ist langsam. Im Urin wird ein erhöhter Gehalt an Heparansulfat und Chondroitinsulfat festgestellt; diese Substanzen kommen jedoch in geringeren Mengen vor. Die Vererbung erfolgt X-chromosomal-rezessiv.
- Typ III, auch als Sanfilippo-Syndrom bezeichnet: Für diesen Typ sind schwere geistige Entwicklungsverzögerungen sowie ein hoher Gehalt an Heparansulfat im Urin charakteristisch. Die Vererbung erfolgt autosomal-rezessiv.
- Typ IV oder das Morquio-Syndrom: Eine wesentliche Skelettdeformierung, insbesondere des Brustkorbs, ist zu beobachten. Im Unterschied zu anderen Typen zeichnet sich diese Form durch die Abwesenheit einer Hornhauttrübung sowie durch fehlende Intellektstörung und ohne grobe Fehlbildungen der Person aus.
- Typ V (Scheie-Syndrom): Die Erkrankung ist durch Hornhauttrübungen und mäßige Skelettdeformitäten gekennzeichnet; die Vererbung erfolgt autosomal-rezessiv.
- Typ VI (Maroto-Lami-Syndrom): Charakteristisch sind eine Wachstumsverzögerung, ein tonnenförmiger Brustkorb sowie grobe Stigmata.
- Typ VII: Die Erkrankung entsteht durch einen Mangel an Beta-Glucuronidase.
Diagnostik der Mukopolysaccharidose.
Die Diagnostik erfolgt durch genealogische, klinische und biochemische Untersuchungen.
Therapie der Mukopolysaccharidose.
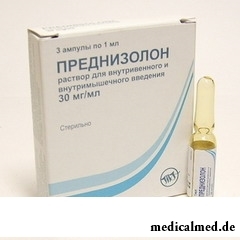
Die Therapie erfolgt vorwiegend symptomatisch; dem Patienten werden Hormonpräparate (Prednisolon, Thyroxin, ACTH) zur Unterdrückung des Polysaccharidgehalts verabreicht. Bei der Behandlung kommen zudem hohe Dosen von Wachstumshormonen sowie Präparate zur Normalisierung der Herzfunktion zum Einsatz. Falls notwendig, können Zytostatika verordnet werden.
Prophylaxe der Mukopolysaccharidose.
Es gibt keine spezifische Prophylaxe; notwendig ist eine pränatale Diagnostik zur Erkennung des Enzymdefekts mittels Amniozentese.
Studien zeigen, dass Frauen mit wöchentlichem Bier- oder Weinkonsum ein erhöhtes Brustkrebsrisiko haben.

Man sollte nicht nur eine schöne Figur anstreben; die neueste Empfehlung lautet: Um abzunehmen, muss man fettreiche Nahrung zu sich nehmen.
Abteilung: Diashow
Viele Eltern von Kindern stoßen im Alter zwischen zwei und vier Jahren auf ein übermäßig launisches Verhalten des Kindes. Das Kind leidet unter ständiger Weinen und Launen nicht nur der Eltern, sondern auch selbst. Worin liegen die Gründe für Kinderlaunen? Wie kann man damit umgehen?
Abteilung: Diashow
Ein Schlaganfall ist eine der häufigsten Erkrankungen des Menschen; jährlich werden weltweit etwa sechs Millionen Fälle dieser Pathologie registriert. Nach Angaben der medizinischen Statistik ereignen sich Schlaganfälle fast dreimal so oft wie Herzinfarkte. Die Erkrankung verläuft schwerwiegend und hat das unangenehme Ergebnis: Die Sterblichkeit liegt bei bis zu 40 % bei Frauen und 25 % bei Männern. Ein großer Teil der Patienten, die einen Schlaganfall erlitten haben, kann nicht vollständig wiederhergestellt werden. Wir bieten den Lesern an, sich damit vertraut zu machen.
Abteilung: Artikel über Gesundheit
Sklera und Bindehaut des Auges werden über eine intensive Blutversorgung mit den nervösen Strukturen des Organs versorgt...
Abteilung: Artikel über Gesundheit
Ayurveda ist eine uralte indische Praxis, die das Lernen umfasst, um physische, psychische und moralische Gesundheit aufrechtzuerhalten; dazu gehören Diäten, Organismusreinigung und Atemübungen.
Abteilung: Artikel über Gesundheit
Trotz der weit verbreiteten Annahme, dass Multiple Sklerose weder durch sklerotische Gefäßwandveränderungen noch durch Gedächtnisstörungen und Konzentrationsstörungen verursacht wird, handelt es sich bei dieser Erkrankung um eine Autoimmunerkrankung. Der pathologische Prozess manifestiert sich in der Degeneration der nervösen Struktur sowie in der Zerstörung ihrer äußeren Hülle – des Myelins. Als Folge des Leidens treten multiple Entzündungen im Zentralnervensystem auf, begleitet von Sehstörungen und rascher Ermüdbarkeit.
Abteilung: Artikel über Gesundheit
Leider müssen wir fast jährlich mit Influenza konfrontiert werden. Es scheint, als wäre diese häufige Erkrankung harmlos.
Abteilung: Artikel über Gesundheit
Jeder von uns hat mehrfach bemerkt, dass Menschen mit gleichem Geburtsalter manchmal völlig unterschiedlich aussehen: Während einer im Alter von 40 bis 45 Jahren bereits wie ein Greis wirkt, ist der andere in 60 noch jung, energisch und vital. Dies liegt daran, dass der Zustand des Körpers...
Abteilung: Gesundheitsartikel
Kaffee – das Lieblingsgetränk vieler. In den letzten Jahrzehnten wurde dieses Getränk mehrfach als außerordentlich nützlich und sogar notwendig für den normalen Lebensvorgang erklärt, obwohl es seit langem zu unserer Gewohnheit geworden ist; dennoch existieren nicht wenige Mythen über seine Eigenschaften und Wirkung auf den menschlichen Organismus. Die Leser können heute die am weitesten verbreiteten ähnlichen Täuschungen kennenlernen.
Abteilung: Gesundheitsartikel
Singen ist ein Vergnügen für alle. Kleine Kinder beschäftigen sich gerne mit dem Gesang, ohne dabei besonders auf das Treffen in die Melodie zu achten; Erwachsene hingegen meistens...
Abteilung: Gesundheitsartikel
Epilepsie zählt zu den häufigsten neurologischen Erkrankungen. Eltern von Kindern, die an dieser Krankheit leiden, stoßen häufig auf Gerüchte und Irrtümer, viele davon stammen aus Zeiten des Mittelalters.
Abteilung: Gesundheitsartikel
Es ist wahrscheinlich, dass jeder Mensch von Erkältungen betroffen ist. Schnupfen, Husten und Kopfschmerzen sind jedem bekannt; im Herbst steigt die Zahl der Infektionen. ORVI hat bereits Schulen und Kindergärten erreicht, während sich die Influenza langsam in den Städten ausbreitet...
Abteilung: Gesundheitsartikel
Gesund und gepflegt auszusehen bedeutet nicht nur, dass man die Umgebung gefällt, sondern auch, dass man sich stark, überzeugt und leistungsfähig fühlt.
Abteilung Gesundheit: Artikel zur Gesundheit
Die Heckenrose – eine der am weitesten verbreiteten dekorativen und medizinischen Pflanzen, die auf dem gesamten Territorium unseres Landes wächst. Für die Mehrheit der Russen ist diese Pflanze vor allem als Quelle für außerordentlich vitaminreiche Früchte bekannt.
Abteilung Gesundheit: Artikel zur Gesundheit
Die Augen – einer der verwundbarsten Bereiche des menschlichen Körpers; daher betreffen Altersveränderungen sie in erster Linie. Kann man das jugendliche Aussehen der Augen über lange Jahre bewahren, und welche Verfahren bieten Kosmetiker an, um dieses Ziel zu erreichen? Oder ist die einzige Variante der Verjüngung die chirurgische Operation – die Blepharoplastik? Wir werden uns dieser Frage widmen.
Abteilung Gesundheit: Artikel zur Gesundheit
Der Sommer in Russland verlief dieses Jahr sehr widersprüchlich: Die Regionen waren sowohl unbarmherziger Hitze als auch Regengüssen und anderen Wetterlagen ausgesetzt...
Abteilung Gesundheit: Artikel zur Gesundheit
Das bekannte Sprichwort ‚Bewahren Sie die Männer!‘ ist nicht auf der leeren Stelle entstanden. In einem bestimmten Sinne hat die Natur die Vertreter des starken Geschlechts weniger für lebenswichtige Unordnung geschaffen, als es anscheinend scheint. Nach Statistiken leiden Männer öfter...
Abteilung Gesundheit: Artikel zur Gesundheit
Der menschliche Körper besteht zu fast 60 % aus Wasser. Diese Flüssigkeit ist für das normale Funktionieren des Organismus von entscheidender Bedeutung: Bereits ein Verlust von anderthalb Prozent führt zu unangenehmen Folgen. Auch bei gesunden Menschen können Wassermangel-Probleme auftreten, wenn sie einige Stunden in der prallen Sonne verbringen und kein Getränk aufnehmen; in diesem Fall lässt sich das Befinden jedoch leicht korrigieren. Viel komplizierter gestaltet es sich hingegen, die Folgen anderer Ursachen zu minimieren...
Abteilung: Artikel über Gesundheit
Antibiotika – Verbindungen, die das Wachstum von Bakterien hemmen – gelten als medizinischer Durchbruch, der Leben retten kann.
Abteilung: Artikel über Gesundheit
Zu den Auswirkungen des Fastens auf den Organismus liegen zahlreiche Aussagen zu Vor- und Nachteilen vor. Es wird angenommen, dass das Kurzzeitfasten (Monodiät) nützlich ist und zur effektiven Ausscheidung von Schlacken beiträgt, während unregelmäßiges Fasten schädlich sein kann.
Abteilung: Artikel über Gesundheit
Phobie – eine störende Angst vor einem bestimmten Inhalt, die wider den Willen des Menschen in einer konkreten Situation auftritt. Die Begriffe Phobie und Angst sind ähnlich; doch während Angst eine physiologische Abwehrfunktion der Psyche darstellt, ist die Phobie davon abweichend. So kann ein Mensch unbewusste, grundlose Ängste (z. B. Zittern, Schüttelfrost), die neurotischen Symptomen entsprechen, vor alltäglichen Ereignissen wie einer Fahrt in der U-Bahn oder dem Anblick eines Hundes nicht ertragen.
Abteilung: Artikel über Gesundheit
Präparate zur Hemmung des Wachstums pathogener Mikroorganismen werden in der klinischen Praxis weit verbreitet eingesetzt.
Abteilung: Artikel über Gesundheit
Die Urlaubszeit ist eingetroffen. Viele Menschen träumen von Erholung in der Natur, touristischen Ausflügen und schönen Stränden. In dieser Zeit möchte man nicht an Gesundheitsprobleme denken; doch andere unangenehme Themen rücken in den Fokus. Der Sommer...
Abteilung: Artikel zur Gesundheit
Die unmittelbare Ursache von Hautkrebs ist unter heutigen Wissenschaftlern nicht einheitlich geklärt; lediglich die Faktoren, die zur Entstehung dieser Erkrankung beitragen, sind exakt definiert. Zu diesen Faktoren zählen: langfristige Einwirkung ultravioletter Strahlung auf die Haut, radioaktive Bestrahlung, thermische Traumata, Verletzungen der Haut durch aggressive chemische Substanzen (Harze, Säuren, Laugen usw.) sowie erbliche Disposition (Vorhandensein von bösartigen Neubildungen in der familiären Vorgeschichte) und...
Abteilung: Artikel zur Gesundheit
Es ist unbestritten, dass unser Land eines der alkoholstärksten Länder der Welt ist. Bei klarem Verständnis, dass der Konsum fester...
Abteilung: Artikel zur Gesundheit
Die Zahl der Langleber ist sehr gering. Bis zum Alter von 90 Jahren erlebt eine Person nur aus 5.000, und die Grenze von 100 Jahren überschreiten lediglich 20.000. Zudem betonen Ärzte, dass jeder von uns sein Schicksal vollständig beeinflussen kann. Dabei geht es um...
Abteilung: Artikel zur Gesundheit
Nach Statistiken leiden bis zum 40. Lebensjahr 25–30 % der Frauen an einer Blasenentzündung. Mit zunehmendem Alter steigt dieser Wert; viele werden in die Statistik nicht einbezogen, da sie sich aus Scham oder anderen Gründen nicht an den Arzt wenden. Bedauerlicherweise kehrt bei der Hälfte der Frauen trotz regelmäßiger Arztbesuche, Langzeitgabe von Antibiotika und eines Lebens im Regime "sich" die Blasenentzündung zurück. Die Symptome einer Blasenentzündung sind eindeutig und lassen sich kaum verwirren: Schmerzen in der Harnblase, Harnbrennen, häufiger Harndrang...
Abteilung: Artikel zur Gesundheit