





Wilson-Konovalow-Syndrom
Allgemeine Charakteristik des Syndroms
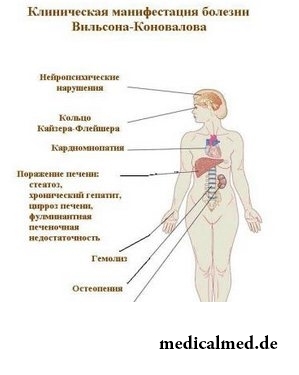
Das Wilson-Konovalow-Syndrom, auch als hepatozelluläre Dystrophie oder hepatotenzikuläre Degeneration bezeichnet, zählt zu den seltenen und schweren genetischen Erkrankungen. Es wird nach dem autosomal-rezessiven Typ vererbt. Dies bedeutet, dass beide Elternteile des Kindes das mutierte Gen ATP7B auf dem 13. Chromosom tragen müssen.
Das Wilson-Konovalow-Syndrom tritt im Kindes- oder Jugendalter auf und wird durch die Ansammlung von Kupfer im Organismus charakterisiert. Dies führt zu einer Leberinsuffizienz sowie zur Entwicklung verschiedener neuropsychischer Störungen.
Es gibt Formen des Wilson-Konovalow-Syndroms mit überwiegend hepatischer, zentralnervöser oder gemischter Symptomatik. Die Lebersymptome treten beim Patienten meist im Alter von 10 bis 15 Jahren auf, während die neurologischen Symptome etwa näher zum 20. Lebensjahr entwickeln.
Das Wilson-Konovalow-Syndrom tritt durchschnittlich bei drei Fällen pro 100.000 Menschen auf. Die Häufigkeit von Trägern des mutierten Gens, das diese Erkrankung verursacht, liegt in der Weltbevölkerung bei etwa 0,6 %. Eine erhöhte Wahrscheinlichkeit für eine Vererbung beider Eltern besteht bei Blutsverwandtschaften.
Symptome des Wilson-Konovalow-Syndroms
Bei der Wilson-Konovalow-Krankheit können zwei Stadien unterschieden werden: das latente Stadium und das Stadium der klinischen Manifestation. Bis zum fünften Lebensjahr zeigt die Erkrankung in der Regel keine Symptome, während bei Kindern ab dem siebten Lebensjahr vor allem die Leberenzyme – insbesondere die Transaminasen – signifikant erhöht sind.
Das Anfangsstadium der Wilson-Konovalow-Krankheit kann akut verlaufen und manifestiert sich durch Fieber, ein asthenisches Syndrom sowie ausgeprägte Hautveränderungen, die auf einen Kupferüberschuss im Körper zurückzuführen sind. Der Krankheitsverlauf schreitet fort bis zur Steatose (Fettansammlung in der Leber) und zu motorischen Störungen.
Die neuropsychiatrischen Erscheinungsformen der Wilson-Konovalow-Krankheit hängen mit den Kupferablagerungen im Körper zusammen. Zu ihnen gehören Sprachstörungen, eine schwache Mimik, vermehrte Speichelabsonderung, Tremor sowie Bewegungs Koordinationsstörungen. Der Intellekt bleibt bei dieser Krankheit vollständig erhalten; jedoch werden beim Patienten Impulsivität, aggressive Reaktionen und Phobien beobachtet.
Neben der Leber sind bei der Wilson-Konovalow-Krankheit auch Nieren, Herz, Knochen und Gelenke betroffen. Im Endstadium der Erkrankung führt das Fehlen einer adäquaten Behandlung zum Tod des Patienten.
Die Diagnostik der Wilson-Konovalow-Krankheit.
Als erste Stufe der Diagnostik dient die klinische Untersuchung. Bereits hier zeigt sich das für diese Krankheit typische Symptom: Das Leberschleimhaut-Symptom (Kupferring). Es handelt sich um eine gelbbraune Verfärbung des Randes der Hornhaut des Auges.
Die nächste Stufe der Diagnostik umfasst laborchemische Untersuchungen von Blut und Urin. Die vorläufige Diagnose wird durch den Nachweis erhöhter Spiegel an Kupfer-bindenden Proteinen sowie die Bestimmung der täglichen Kupferausscheidung im Urin bestätigt.
Zu den instrumentellen diagnostischen Verfahren gehören Ultraschall, MRT und CT. Hier werden Hepatomegalie und Splenomegalie sichtbarisiert sowie eine Zerstörung von Nervenzellknoten im Gehirn nachgewiesen.
Der genetische Bereich der Diagnostik umfasst die Genotypisierung des Patienten und seiner nächsten Verwandten zur Identifizierung des pathologischen Gens.
Die Behandlung der Wilson-Konovalow-Krankheit.

Die Therapie der Wilson-Konovalow-Krankheit ist symptomatisch. Ihr Ziel besteht in der Reduktion der im Körper vorhandenen Kupfermenge sowie der Senkung bereits bestehender Kupferspeicher.
Ein wichtiger Bereich der Behandlung der Wilson-Krankheit ist die lebenslange Diät mit vollständiger Exklusion von kupferhaltigen Lebensmitteln wie Bohnen, Kaffee und Schokolade.
Die Pharmakotherapie der Wilson-Krankheit erfolgt durch Präparate, die Kupfer aus dem Organismus entfernen; hierfür werden D-Penicillamin sowie Zinksalze eingesetzt, wobei das Dosierungsschema individuell angepasst wird.
Ein wesentlicher Bestandteil der Therapie sind regelmäßige prophylaktische Untersuchungen alle 6 bis 12 Monate zur Kontrolle der Behandlungseffektivität durch physikalische Verfahren sowie Blut- und Urintests unter Überwachung von Leber- und Nierenfunktionen.
Bei schwerer Organschädigung wird die Wilson-Krankheit chirurgisch behandelt; eine Lebertransplantation ermöglicht dem Patienten bei erfolgreichem Eingriff eine vollständige Genesung ohne lebenslange Therapie.
Die durchschnittliche Lebenserwartung von Linkshändern liegt niedriger als die von Rechtshändern.

Lange Zeit galten Antibiotika als Allheilmittel gegen jede Krankheit und wurden selbst bei unbedeutenden Infektionszeichen verschrieben, auch bei Seuchen.
Rubrik: Artikel zur Gesundheit
Ein gesundes und gepflegtes Aussehen bedeutet nicht nur, die Umgebung zu gefallen, sondern auch, sich stark, überzeugt und vital zu fühlen. Experten im Bereich der Kosmetik weisen häufig darauf hin, dass fast alle Frauen verstehen, wie wichtig es ist, für die Haut zu sorgen...
Rubrik: Artikel zur Gesundheit
Gesundheit und Attraktivität sind ewige Werte, die Menschen oft dazu verleiten, ungewöhnlichste Inhaltsstoffe und Techniken einzusetzen. Lassen Sie uns elf außergewöhnliche und manchmal nicht angenehme Schönheitsprozeduren betrachten, auf die sich der Mensch in seinem Streben nach Schönheit und Jugend einlässt...
Rubrik: Artikel zur Gesundheit
Die bekannte Losung ‚Bewahren Sie die Männer!‘ ist nicht aus dem Nichts entstanden. In gewissem Sinne hat die Natur die Vertreter des Starken geschaffen...
Rubrik: Artikel zur Gesundheit
Die Bedeutung der Nieren für den Organismus ist nicht zu unterschätzen. Diese Organe reinigen das Blut von Abbauprodukten der Nahrung und scheiden überschüssige Flüssigkeit aus, gleichzeitig sind sie an der Produktion bestimmter Hormone beteiligt, die für den normalen Zustand des Körpers unerlässlich sind.
Rubrik: Artikel zur Gesundheit
Erkältung – ein geläufiger Krankheitszustand, begleitet von Schnupfen, Husten, hohem Fieber und Halsbeschwerden. Oft beginnen wir mit der Hoffnung auf Genesung, Medikamente einzunehmen; diese sind jedoch nicht immer harmlos. Natürliche Mittel können die Krankheitserscheinungen hingegen leicht lindern: Sie entfernen nicht nur die Symptome, sondern versorgen den geschwächten Organismus zudem mit nützlichen Substanzen. Wir stellen Ihnen acht Getränke vor, die erfolgreich eingesetzt werden können.
Rubrik: Artikel über Gesundheit
Fehlt der Appetit und ist eine vollständige Ernährung unmöglich, kommen Perkussionen – also kleine Portionen – zur Hilfe...
Rubrik: Artikel über Gesundheit
Viele Eltern stoßen bei Kindern im Alter von zwei bis vier Jahren auf übermäßiges launisches Verhalten. Das Kind leidet nicht nur durch ständiges Weinen und Launen, sondern auch selbst darunter. Worin liegen die Gründe für Kinderlaunen? Wie kann man damit umgehen?
Rubrik: Slideshow
Instabilität des Knöchelgelenks ist ein sehr verbreitetes Problem. Ihr Vorhandensein zeigt sich in der Neigung zu Fußfehlstellungen beim Gehen auf den Absätzen, häufigen krankhaften Dehnungen der Bänder sowie schmerzhaften und namenlosen Schmerzen der Finger des Fußes selbst nach geringsten Belastungen. Gewöhnlich nehmen Menschen mit solcher Pathologie unangenehme Empfindungen durch schmerzlindernde Salben oder Rastrike ab, doch dies führt nicht zur radikalen Lösung des Problems. Stattdessen kann man bei bekannter Beharrlichkeit das obere Sprunggelenk im Haus festigen.
Rubrik: Artikel über Gesundheit
Der Kauf von Medikamenten in Moskau stellt ein Problem dar: Apotheken gibt es nicht mehr; bei jedem Bewohner der Hauptstadt fehlt sie schrittweise.
Rubrik: Artikel über Gesundheit
Für viele Frauen klingt der Begriff 'Fett' wie ein negatives Urteil. Im Streben nach idealer Statur versuchen sie vor allem, alle Platten aus dem Menü auszuschließen, die Fette enthalten, ohne jedoch eine klare Vorstellung von der Rolle dieser Substanzen in den Stoffwechselprozessen zu haben.
Die Abteilung: Artikel zur Gesundheit
Die Medizin entwickelt sich rasant: Was vor kurzem noch als Wunder galt, ist heute selbstverständlich. Wir wundern uns nicht mehr darüber, dass Menschen mit künstlichen Gelenken und Gliedmaßen durch Sport aktiv werden können, dass Organtransplantationen zum Standard wurden und neue Krebsmedikamente die Sterblichkeit um ein Vielfaches senken. Die Leistungen der plastischen Chirurgie, dank denen Menschen auch im Alter von 60 Jahren noch für Schönheit und Frische sorgen, sind längst keine Sensation mehr.
Die Abteilung: Artikel zur Gesundheit
Genetisch veränderte Organismen (GVO) sind Pflanzen oder Tiere (meist landwirtschaftlich), deren Genotyp...
Die Abteilung: Artikel zur Gesundheit
Das Klimakterium ist der Prozess des Erlöschens der Fortpflanzungsfunktion im Rahmen des Alterns. Ein Hauptmerkmal seines Eintritts bei Frauen ist die Unterbrechung des Menstruationszyklus. Offiziell wird die Menopause diagnostiziert, wenn in sechs aufeinanderfolgenden Monaten keine Menstruationen beobachtet werden...
Die Abteilung: Artikel zur Gesundheit
Für die meisten Erwerbstätigen stellt das Problem der Zwischenmahlzeiten eine große Herausforderung dar. Die Frage, ob früh oder spät gegessen wird – also zwischen Frühstück und Mittagessen oder zwischen Mittagessen und Feierabend –, ist entscheidend: Wie kann man sich energetisch auffüllen, ohne den Organismus durch schädliche Komponenten oder überflüssige Kalorien zu belasten? Wir bieten Ihnen ein Verzeichnis von Lebensmitteln an, das diesen Anforderungen vollständig entspricht.
Die Abteilung: Artikel zur Gesundheit
Vitaminpräparate gehören zweifellos zu den beliebtesten Arzneimitteln; kaum ein Mensch hat noch nicht davon gehört.
Rubrik: Artikel zur Gesundheit.
Die extrakorporale Befruchtung zählt zu den modernsten Methoden der Unfruchtbaritätsbekämpfung und hat bereits vielen Paaren geholfen, Eltern zu werden; die Prozedur ist jedoch komplex und aufwendig.
Rubrik: Artikel zur Gesundheit.
Etwa 20 % der Bevölkerung unseres Planeten leiden unter Hypertonie (anhaltender Erhöhung des arteriellen Blutdrucks). Diese Erkrankung verschlechtert den Lebensstandard, mindert die Arbeitsfähigkeit und bedroht bei fehlender systematischer Behandlung mit schwerwiegenden Komplikationen wie Herzinfarkt oder Schlaganfall, die zu Behinderung oder plötzlichem Tod führen können. Die meisten Patienten werden von Ärzten zur Aufrechterhaltung eines akzeptablen Blutdrucks medikamentös behandelt.
Rubrik: Artikel zur Gesundheit.
Sind Sie Büroangestellter, Fahrer oder Wintersportler? Oder träumen Sie von einem Leben ohne Fahrrad? In diesem Fall führen Sie einen bewegungsarmen Lebensstil.
Rubrik: Artikel zur Gesundheit.
Die Anwendung der Hypnose wird seit zwei Jahrtausenden praktiziert. In dieser Zeit haben Wissenschaftler viel über das Phänomen gelernt und nutzen sie, um den Zustand schwer leidender Patienten zu lindern.
Rubrik: Artikel zur Gesundheit.
Der Gedanke, dass die Körpermasse viel zu groß ist, besucht mindestens einmal im Leben 80 bis 95 % der Frauen. Viele sind von dieser Idee so besessen, dass sie ständig nach neuen Wegen zur Gewichtsreduktion suchen. Das Spektrum dieser Methoden ist unübersichtlich, und einige sind gesundheitsschädlich.
Rubrik: Artikel über Gesundheit
Unter heutigen Wissenschaftlern herrscht keine einheitliche Meinung bezüglich der unmittelbaren Ursache für Hautkrebs. Die genauen Mechanismen sind noch nicht vollständig geklärt.
Rubrik: Artikel über Gesundheit
Hersteller von Milchersatzprodukten für Kinder versichern, dass diese Mischungen ideal auf die Bedürfnisse von Säuglingen abgestimmt sind. Wenn eine Mutter aufgrund schwerwiegender Gesundheitsprobleme künstlich ernähren muss, können diese Ersatzprodukte eine sinnvolle Alternative sein.
Rubrik: Artikel über Gesundheit
Die Cellulitis ist ein weit verbreiteter kosmetischer Defekt, der bei Frauen früh oder etwa bei 80 % später auftritt. Sein Auftreten hängt mit strukturellen Veränderungen in der subkutanen Fettschicht zusammen. Zuerst zeigen sich auf der Hautoberfläche Unebenheiten (Wölbungen und Einbuchtungen), gefolgt von kleinen Verdichtungen, was den sogenannten „Apfel-Haut-Effekt" erzeugt. Diese Veränderungen im Zustand des subkutanen Gewebes deuten auf eine hormonelle Dysbalance im Körper hin.
Rubrik: Artikel über Gesundheit
Neujahr, Hochzeit und Geburtstag sind Anlässe, bei denen der russische Mensch traditionell Alkohol konsumiert. Es gibt verschiedene Strategien, um die negativen Auswirkungen zu minimieren.
Rubrik: Artikel über Gesundheit
Der Begriff "Kleber" bezeichnet eine Gruppe von Proteinen (Gluten), die in Roggen, Gerste und Weizen vorkommen. Für die Mehrheit der Menschen ist der Verzehr von Lebensmitteln mit Klebstoff nicht nur sicher, sondern auch sehr nützlich. Dennoch existiert eine ganze Reihe von Mythen.
Rubrik: Artikel zur Gesundheit
Der Tee wird fast allen empfohlen. Dieses Getränk wirkt belebend und enthält Gerbstoffe, die die Aktivität krankheitserregender Mikroorganismen unterdrücken können. In letzter Zeit haben Tees mit pflanzlichen Zusätzen große Popularität erlangt. Heilkräuter, Gewürze und Früchte, die solche Mischungen bilden, bereichern das Getränk um Vitamine und Spurenelemente; dadurch wird der Nährwert erhöht und ein zusätzlicher therapeutischer Effekt erzielt.
Rubrik: Artikel zur Gesundheit