





Glykogenosen
Glykogenspeicherkrankheiten sind Sammelbegriff für eine Gruppe erblicher Störungen, die durch Defekte oder das Fehlen von Enzymen hervorgerufen werden, die am Abbau oder der Synthese des Glykogens beteiligt sind.
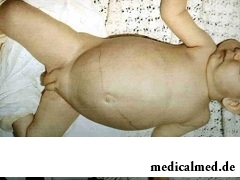
Das Glykogen dient als Energiereserve und versorgt den Organismus bei Bedarf sofort mit Glukose. Wird es in der Leber abgebaut, stellt es eine kontinuierliche Energiequelle für Erythrozyten und das Gehirn dar. Bei Glykogenspeicherkrankheiten wird die normale Funktion der Enzyme gestört; im Ergebnis lagern sich entweder zu viel Glykogen oder pathologische Formen davon im Organismus an.
Glykogenspeicherkrankheiten sind Sammelbegriff für eine Gruppe erblicher Störungen, die durch Defekte oder das Fehlen von Enzymen hervorgerufen werden, die am Abbau oder der Synthese des Glykogens beteiligt sind. Diese Erkrankungen stellen eine kontinuierliche Energiequelle für Erythrozyten und das Gehirn dar. Bei Glykogenosen wird die normale Funktion der Enzyme gestört; im Ergebnis lagern sich entweder zu viel Glykogen oder pathologische Formen davon im Organismus an.
Die verschiedenen Formen der Glykogenose unterscheiden sich primär durch das betroffene Enzym sowie den Ort des Glykogenspeichers. Man unterscheidet zwischen Typen, die eine Störung der Glykogensynthese (z. B. bei der Glykogenose-Typ IV) oder des Glykogenabbaus (z. B. bei den klassischen Speicherkrankheiten wie Typ I bis III) verursachen.
Je nach Lokalisation des Glykogenspeichers in Organen und Geweben werden die Glykogenosen in folgende Hauptformen eingeteilt:
- Hepatozytose;
- Myozytose;
- Die generalisierende Form.
Merkmale der Pechenot-Form: Die Formen der Glykogenspeicherung treten gewöhnlich im 8.–9. Lebensmonat auf. Zu Beginn manifestieren sich seltene Attacken einer Hyperglykämie, die von Krämpfen in den Gliedmaßen und einem vorübergehenden Bewusstseinsverlust begleitet werden. Diese Attacken ereignen sich am Morgen bis zum Beginn der Mahlzeit und lassen sich durch die Aufnahme von süßem Wasser verhindern. Die Kinder weisen ein spezifisches Aussehen auf: unnatürlich dünne Hände und Beine, einen großen Bauch, eine Größenverzögerung (das Kind wirkt wie eine Puppe) sowie eine optisch auffällige Hepatomegalie. In den ersten 4–5 Lebensjahren ist der Verlauf am gefährlichsten, da der Ablauf durch verschiedene Infektionskrankheiten erschwert wird. Mit zunehmendem Alter entwickeln sich im Organismus Kompensationsmechanismen des Stoffwechsels, die zu einer wesentlichen Verbesserung des Zustands beitragen. In der Regel hat die Glykogenose keinen Einfluss auf die intellektuelle Entwicklung.
Merkmale der Muskelform: Die Ansammlung von Glykogen zeigt sich gewöhnlich im Alter von 7–10 Jahren. Das kranke Kind ermüdet bei geringen körperlichen Belastungen schnell und wird bewegungsarm. Die fortschreitende Muskelschwäche provoziert Schmerzen in den belasteten Muskeln, eine Verstärkung des Herzklopfens sowie Atemnot. Diese Symptome entwickeln sich bis zum 25.–35. Lebensjahr; auf dem Aussehen der Patientinnen spiegelt dies jedoch nicht wider, da Leber und Nieren gewöhnlich im Normbereich liegen. Die Veränderungen zeigen sich lediglich in der Muskeltextur, für die Atrophie und Hypotonie charakteristisch sind.
Bei der generalisierten Form der Glykogenose lagert sich tatsächlich eine große Menge an Glykogen in allen Geweben und Organen ab. Das klinische Bild ist vielfältig, der Verlauf fortschreitend.
Aglykogenose.
Aglykogenose oder Glykogenose des Nulltyps: eine erbliche genetische Erkrankung, die durch das vollständige Fehlen der Uridindiphosphat-Glukose-Glykogen-Transferase (oder Glykogensynthetase) im Organismus des Patienten hervorgerufen wird. Dies ist das Enzym, das für die Synthese von Glykogen verantwortlich ist.
Die Aglykogenose geht – ebenso wie die klassische Glykogenose – mit Hypoglykämien und hypoglykämischen Krämpfen einher (vorzugsweise am Morgen). Dabei kann der Blutzuckerspiegel auf Werte zwischen 7–12 mg % absinken. Diese Symptome können durch außerordentlich häufige nächtliche Fütterungen verhindert werden. Zur Bestätigung der Aglykogenose ist eine biopsische Untersuchung der Leber auf Enzyme und Glykogen erforderlich.
Die Behandlung der Aglykogenose bzw. der Glykogenose Typ 0 ist rein symptomatisch, und die Prognose des Krankheitsverlaufs ist leider ungünstig.
In Familien mit Vorerkrankungen dieser Art ist eine medizinisch-genetische Beratung in frühen Schwangerschaftsstadien zur Verhinderung einer Geburt eines Kindes mit Aglykogenose oder Glykogenose Typ 0 erforderlich.
Die Behandlung der Glykogenosen und der Aglykogenose.
Zu den Hauptzielen der Therapie bei verschiedenen Formen der Glykogenose gehört die Verhinderung von Hypoglykämien. Zur Senkung des Blutzuckerspiegels wird empfohlen:
- Eine proteinreiche Ernährung
- Häufige Nahrungsaufnahme
- Aufnahme von Fruktose, Multivitaminierten und ATP
- Substitution fehlender Enzyme je nach Form der Glykogenose
- Reduktion körperlicher Belastungen.
Patientinnen mit verschiedenen Formen der Glykogenosen sollten zwingend eine ambulante Betreuung durch einen Internisten (Kinderarzt) sowie Ärzte eines medizinisch-genetischen Zentrums erhalten.
Bei 5 % der Patientinnen kann das Antidepressivum Klomipramin zu einem Orgasmus führen.

Nach Angaben der Ärzte leiden etwa die Hälfte der Männer im Alter zwischen 25 und 50 Jahren an Verwirrungen des Gehirns, wenden sich jedoch oft nicht an einen Arzt.
Abteilung: Artikel über Gesundheit.
Unter Subfebrilität versteht man eine Erhöhung der Körpertemperatur auf Werte bis zu 38 Grad; liegt diese Temperatur über drei Tage an, ohne dass häufig sichtbare Gründe vorliegen, handelt es sich um ein offenkundiges Zeichen für Störungen im Organismus.
Abteilung: Artikel über Gesundheit.
Das Baden mit Heilfarben und Pflanzen (Phyto-Bäder) war bereits seit der Zeit von Kleopatra verbreitet; heute sind Phyto-Bäder ein einfaches und verfügbares Mittel, das nicht nur zur Linderung nervöser Anstrengung dient, sondern auch bei der Behandlung vieler Krankheiten eingesetzt werden kann. Besonders ergebnisreich sind Grasbäder bei der Behandlung von Erkältungen, Osteochondrose, Radikulitis, Hautkrankheiten sowie Erkrankungen des Bewegungsapparates und der Gefäße.
Abteilung: Artikel über Gesundheit.
Mit Beginn der Urlaubszeit träumen viele Russen von Erholung in der Natur; touristische Ausflüge und die Nutzung schöner Strände werden bereits erwartet.
Abteilung: Artikel zur Gesundheit
Das Gedächtnis ist eine Fähigkeit des Zentralnervensystems zur Speicherung von Informationen und bedarfsgerechte Wiedergabe; der Mechanismus dieses Prozesses bis zum Ende ist noch nicht vollständig erforscht.
Abteilung: Artikel zur Gesundheit
Der kurz nach der Geburt geborene Mensch ist von der Liebe seiner erwachsenen Familienmitglieder sowie deren Sorge umgeben; ohne diese Unterstützung kann das Kind nicht bestehen. Manche Eltern glauben fälschlicherweise, dass zarte Fürsorge und Liebkosung ausreichen, damit sich das Kind richtig entwickelt und glücklich wird, doch dies ist nicht der Fall. Es ist entscheidend, die Besonderheiten des Wachstums, die Gründe für das Verhalten und mögliche Probleme frühzeitig zu erkennen. Nur eine "sehende" Liebe kann dem kleinen Menschen garantieren, was er wirklich benötigt.
Abteilung: Artikel zur Gesundheit
Hohes Fieber ist ein häufiges Symptom weit verbreiteter Erkrankungen wie Infektionen der oberen Atemwege (ORWI), Angina pectoris oder Pneumonie; um die Körpertemperatur zu senken...
Abteilung: Artikel zur Gesundheit
Herpes Simplex Typ 1 (eine Infektionskrankheit, die sich durch periodisch auftretende Bläschen auf den Lippen äußert) zählt zu den häufigsten Leiden. Nach Statistiken sind nur etwa 5 % der Bevölkerung gegen diesen Erreger immun, und...
Abteilung: Artikel zur Gesundheit
Gelenkerkrankungen beginnen für den Menschen oft unauffällig. Die frühen Stadien des Knorpelabbaus, die das weiche und reibungslose Gleiten der Knochenköpfe in den Gelenkkapseln ermöglichen, verlaufen langsam und schmerzfrei. Besonders bedenklich ist, dass dieser Prozess nicht zwingend mit fortgeschrittenem Alter verbunden sein muss: eine Degeneration der Gelenkflächen kann bereits nach dem 30. Lebensjahr auftreten. Dies bedeutet, dass jeder arbeitsfähige Mensch jederzeit praktisch auf traurige Folgen stoßen kann.
Abteilung: Artikel über Gesundheit
Heute sind neben 30 Erkrankungen bekannt, die sexuell übertragen werden; dem breiten Verbreitung dieser Leiden wird außerordentlich viel Aufmerksamkeit geschenkt.
Abteilung: Artikel über Gesundheit
Für die Mehrheit der Beschäftigten ist das Übergewicht ein drängendes Problem. Die Frage, ob man zwischen Frühstück und Mittag oder zwischen Mittag und Feierabend schnell essen kann, um energetisch bedingt zu sein, ohne jedoch eine Überlastung zu erleiden, stellt sich früh oder spät.
Abteilung: Artikel über Gesundheit
Die Hirnleistung ist außerordentlich komplex und in vieler Hinsicht noch nicht vollständig erforscht. Auch die Besonderheiten der Denkprozesse, die während des Schlafes auftreten, bestätigen dies. Wir werden Ihnen einige davon erläutern.
Abteilung: Artikel über Gesundheit
Der Begriff 'Leimstoff' (alternativ auch als Kleber bezeichnet) umfasst die Gruppe der Proteine, aus denen Roggen, Gerste und Weizen bestehen. Für die Mehrheit der Bevölkerung ist dies von Bedeutung.
Abteilung: Artikel über Gesundheit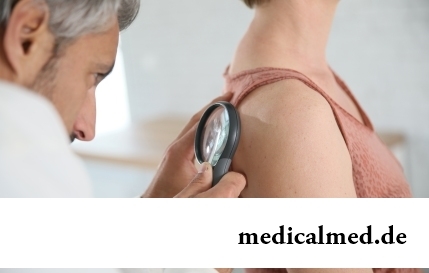
Es herrscht unter den Wissenschaftlern derzeit keine einheitliche Meinung über die unmittelbare Ursache des Entstehens von Hautkrebs. Genau bestimmt sind lediglich die Faktoren, die zur Entwicklung dieser Erkrankung beitragen: die langdauernde Einwirkung ultravioletter Strahlen auf die Haut sowie Radiofluorid.
Abteilung: Artikel zur Gesundheit
Die Lebenserwartung variiert je nach Region. Sie wird beeinflusst durch soziale Stabilität, Wirtschaftswachstum, Ernährungssituation und Versorgungsstandard sowie den Haushaltskomfort. Zudem spielt die Alphabetisierung der Bevölkerung eine Rolle für die Beachtung von Sanitär- und Hygienestandards sowie weiteren Faktoren. Dennoch bleibt ein konstanter Kennwert bestehen: Fast in allen Ländern leben Frauen durchschnittlich 7 bis 10 Jahre länger als Männer. Heute werden wir über die Gründe dieser Erscheinung sprechen.
Abteilung: Artikel zur Gesundheit
Der menschliche Körper besteht zu etwa 60 % aus Wasser. Für das normale Funktionieren des Organismus ist der Flüssigkeitsverlust von entscheidender Bedeutung...
Abteilung: Artikel zur Gesundheit
Aspirin (Acetylsalicylsäure) gehört zu den am weitesten verbreiteten Arzneimitteln. Es ist in fast jeder Hausapotheke vorhanden, und viele greifen bei ersten Anzeichen von Unwohlsein darauf zurück, ohne jedoch eine klare Vorstellung über seine Eigenschaften und therapeutischen Wirkungen zu haben.
Abteilung: Artikel zur Gesundheit
Der Stoffwechsel läuft bei jedem Menschen individuell ab. Dennoch besteht bei allen ein Zusammenhang zwischen der Geschwindigkeit dieses Prozesses und dem Abbau von Übergewicht. Leider neigen die Menschen dazu, zahlreiche vermeintlich "wundertätige" Diäten auszuprobieren; sie berücksichtigen diesen Umstand jedoch häufig nicht und beginnen mit entschlossensten Absichten sich so künstlich zu ernähren, was den Metabolismus bremst statt ihn zu beschleunigen. Dies führt außer der vollständigen Enttäuschung über falsch ausgewählte Systeme oft noch zu weiteren gesundheitlichen Risiken.
Abteilung: Artikel zur Gesundheit
Das endokrine System erfüllt im menschlichen Organismus eine außerordentlich wichtige Rolle; es reguliert praktisch alle Prozesse des Lebensvorgangs.
Abteilung: Artikel über Gesundheit
Eine Erkältung ist ein bekannter Zustand, der von Schnupfen, Husten, hohem Fieber und Halsbeschwerden begleitet wird. Oft greifen wir zunächst zu Medikamenten in der Hoffnung auf Genesung, die jedoch nicht immer unschädlich sind.
Abteilung: Artikel über Gesundheit
Klebsche Enzephalitis ist eine der gefährlichsten Viruserkrankungen, deren Erreger durch blutsaugende Insekten übertragen werden, die auf dem gesamten Territorium unseres Landes vorkommen. Ein von Zecken gebissener Mensch kann sich zudem mit Ehrlichiose, Bartonellose, Babesiose sowie Mykoplasmose infizieren; eine Infektion mit der Lyme-Borreliose ist ebenfalls möglich. Wie bei der Enzephalitis betreffen diese Leiden das Zentralnervensystem. Da keine spezifische virustötende Therapie existiert, ist die Prognose sehr ungünstig.
Abteilung: Artikel über Gesundheit
Die Belege für die Effektivität von Mildronat bei der Behandlung der ischämischen Herzkrankheit (Stenokardie) finden sich in vielen Veröffentlichungen.
Abteilung: Artikel über Gesundheit
Die Anzahl der Langleber ist sehr gering: Bis zum Alter von 90 Jahren erreicht ein Mensch nur etwa 5.000, und lediglich 20.000 Personen überschreiten die Grenze von 100 Jahren. Zudem behaupten manche Ärzte, dass jeder sein eigenes Schicksal voll beeinflussen kann; dabei ist jedoch zu bedenken...
Abteilung: Artikel über Gesundheit
Der Lauf – eine der verfügbaren und ergebnisreichen Methoden zur Stärkung des Organismus. Die Kenntnis seines erstaunlichen Nutzens veranlasste jeden von uns zumindest einmal dazu, selbst wenn es den Lauf zu begehen galt, doch nur wenige praktizieren diese Beschäftigung regelmäßig. Obwohl es beim Jogging scheinen mag (es gibt einen leichten Gesundheitslauf), machen beginnende Läufer oft Fehler, die zur vollen Unterbrechung des Trainings führen. Wir betrachten zehn segensreiche Räte für Neulinge, die ihnen erlauben werden, regelmäßig zu trainieren.
Abteilung: Artikel über Gesundheit
Schwäche des Knöchelgelenks – ein sehr verbreitetes Problem. Ihr Vorhandensein deutet auf eine Neigung zu Fußkrämpfen der Beine hin...
Abteilung: Artikel über Gesundheit
Präparate, die das Wachstum oder den Lebensvorgang pathogener Mikroorganismen unterdrücken, werden seit 40 Jahren des vorigen Jahrhunderts in der klinischen Praxis breit eingesetzt. Ursprünglich wurden Antibiotika nur als natürliche Substanzen (tierischer, pflanzlicher oder mikrobieller Herkunft) bezeichnet...
Abteilung: Artikel über Gesundheit
Gesund und gepflegt auszusehen bedeutet nicht nur, die Umgebung zu gefallen, sondern auch, sich stark, überzeugt und vital zu fühlen. Experten im Bereich der Kosmetologie bemerken häufig, dass viele Frauen verstehen müssen, dass sie für ihre Gesichtshaut sorgen müssen. Viele verwenden kosmetische Mittel falsch oder nutzen verschiedene Verfahren unzureichend, da ihnen genaue Informationen über deren tatsächliche Wirkung fehlen; dies führt nicht zum besten Ergebnis.
Abteilung: Artikel über Gesundheit