





Laterale amyotrophe Sklerose
Die laterale amyotrophe Sklerose ist eine fortschreitende Erkrankung der Motoneurone verschiedener Ätiologie, die sich durch zunehmende Muskelschwäche, Muskelatrophie sowie Spastik der Muskeln auszeichnet. Zudem treten Störungen der Sprache (Dysarthrie), des Schluckens (Dysphagie) und der Atmung auf. Die laterale amyotrophe Sklerose ist die häufigste Erkrankung der Motoneurone. Muskelschwäche und -atrophie entstehen durch Degeneration der oberen und unteren Motoneurone. Muskeln, die nicht mehr funktionieren können und atrophieren, schwächen sich zunehmend; im Endeffekt verlieren sie die Fähigkeit zur Bewegung und Kontrolle aller willkürlichen Bewegungen.
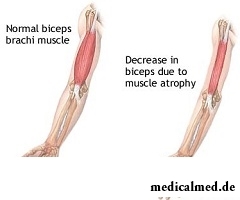
Symptome der lateralen amyotrophen Sklerose
Zu den frühen Symptomen der lateralen Amyotrophen Sklerose gehören in der Regel offensichtliche Schwäche und Muskelatrophie. Weitere Symptome umfassen Muskelzuckungen, Krämpfe, Härte der betroffenen Muskeln sowie undeutliche Sprache. Der Zustand der Körperteile, die von den frühen Symptomen betroffen sind, hängt davon ab, welche Motoneurone im Organismus primär geschädigt wurden.
In etwa 75 % der Fälle beginnt die Muskelatrophie an Händen oder Beinen. Patienten mit Beinatrophy können Unbeholfenheit beim Gehen oder Laufen erleben, Stolpern und Stürze auf den Boden. Patienten mit Handatrophy bemerken Schwierigkeiten bei Bewegungen, die Feinmotorik erfordern – wie das Knöpfen von Kleidungsstücken, das Drehen des Schlüsselrings oder das Drehen des Schlüssels im Schloss. Manchmal beschränken sich die Symptome zunächst auf eine einzelne Gliedmaße über einen längeren Zeitraum oder während der gesamten Krankheitsdauer.
In etwa 25 % der Fälle beginnt die Krankheit mit Schwierigkeiten beim Sprechen oder Schlucken. Die Sprache kann undeutlich und sehr leise werden. Weitere Symptome umfassen Schluckbeschwerden sowie einen Verlust der Beweglichkeit der Zunge.
Bei einem kleineren Anteil der Patienten manifestiert sich das Krankheitsbild zunächst durch eine Atrophie der Interkostalmuskulatur, die für die Atembewegungen verantwortlich ist.
Im Verlauf des Krankheitsverlaufs entwickeln alle betroffenen Patienten die folgenden Symptome der lateralen amyotrophischen Sklerose:
- Zu beachten ist das Problem der Umstellung.
- Schluckstörungen (Dysphagie) sowie Sprach- oder Wortfindungsstörungen (Disarthrie);
- Muskelhärte (Spastizität);
- Erhöhte Reflexbereitschaft (inklusive gesteigerter Brechneigung);
- Muskelkrämpfe;
- Vorübergehende Muskelzuckungen.
Darüber hinaus zeigen 15–45 % der Patienten einen Pseudobulbärismus, das heißt eine gestörte emotionale Beweglichkeit: unkontrolliertes Lachen, heftiges Weinen oder übertriebenes Äußern von Emotionen.
Obwohl die Reihenfolge und das Tempo des Symptomauftretens bei den meisten Patienten variieren – einige können sich selbstständig vom Bett erheben oder die Hände bewegen –, lässt sich der Krankheitsverlauf mittels einer klinischen Skala für laterale Amyotrophie messen, die auf Grundlage eines klinischen Gesprächs aus 12 Fragen abgeleitet wird.
Der Verlauf der Erkrankung ist bei Patienten unter 40 Jahren in der Regel langsamer; meist beschränkt sich die Krankheit auf eine einzelne Gliedmaße.
Zu den Symptomen des späten Stadiums der lateralen Amyotrophie gehören:
- Schluckbeschwerden und Schwierigkeiten beim Kauen von Nahrungsmitteln, die durch Anstrengung sowie das Risiko einer Erstickung oder Aspiration in die Lunge verstärkt werden.
- Schwäche des Zwerchfells und der Zwischenrippenmuskulatur, was die Atmung sowie Lungenfunktionen wie die Vitalkapazität beeinträchtigt und den Blutdruck während der Inspiration senkt;
Die Mehrheit der Patienten mit lateraler amyotrophischer Sklerose stirbt infolge einer respiratorischen Insuffizienz meist innerhalb von drei bis fünf Jahren nach Symptombeginn; das mittlere Überleben beträgt 39 Monate, wobei nur 4 % länger als zehn Jahre leben.

Behandlung der lateralen Amyotrophen Sklerose
Zur Behandlung der lateralen Amyotrophen Sklerose wird Riluzol (Rilutek) eingesetzt. Dies ist das einzige Medikament, das den Krankheitsverlauf verlangsamt und die Lebenszeit um einige Monate verlängert; bei frühen Stadien unterstützt es zudem die Zeit bis zur Notwendigkeit einer Beatmung.
Weitere Behandlungsmethoden der lateralen Amyotrophen Sklerose zielen auf die Linderung von Symptomen und die Verbesserung der Lebensqualität ab. Diese supportive Therapie wird primär durch Allgemeinmediziner sowie spezialisiertes medizinisches Personal gewährleistet, das mobile und komfortable Bedingungen für Patienten sicherstellt.
Ärztliches Personal kann Medikamente verschreiben, um Ermüdung zu reduzieren, Muskelkrämpfe zu schwächen, Spastik zu kontrollieren sowie exzessive Speichelsekretion und Sekretionsstörungen zu mindern. Diese Mittel helfen zudem bei der Linderung von Schmerzen, Depressionen, Schlafstörungen, Dysphagie und Obstipation. Baklofen und Diazepam werden häufig zur Kontrolle von Spastik eingesetzt, die durch laterale Amyotrophe Sklerose hervorgerufen wird, während Trihexyphenidyl oder Amitriptylin verschrieben werden können, wenn Schluckbeschwerden mit Speichelansammlung auftreten.
Es existieren sehr spezifische medizinische Syndrome, beispielsweise das Syndrom des pathologischen Erzählens von Gegenständen. Im Magen einer Patientin, die an dieser Manie litt, wurden 2500 fremde Gegenstände gefunden.

Ein guter Appetit galt stets als Zeichen einer guten Gesundheit; die korrekte Funktion des Mechanismus, der auf das Nahrungserfordernis reagiert...
Abteilung: Artikel über Gesundheit.
Manche Menschen meinen, dass für die Medizin des 21. Jahrhunderts fast keine Geheimnisse im Bereich der menschlichen Gesundheit bestehen; das ist jedoch nicht so. Je mehr Antworten die Gelehrten erhalten, desto komplexere Fragen stellt ihnen das Leben vor. Zudem gibt es Erkrankungen, die sich nicht erklären lassen...
Abteilung: Artikel über Gesundheit.
Wir betonen zunächst, dass ein isoliertes Leiden unter dem Haushaltsnamen „Salzablagerung" nicht existiert; tatsächlich handelt es sich bei dieser Bezeichnung um eine Stoffwechselstörung, die zur Entstehung einer ganzen Reihe von Erkrankungen führt. Der pathologische Prozess besteht darin, dass im Organismus eine Ansammlung von Salzen auftritt (was meist infolge einer Störung des Wasser-Mineralstoffwechsels oder eines unzureichenden Ergebnisses der Ausscheidungsarbeit geschieht)...
Abteilung: Artikel über Gesundheit.
Die Bedeutung der Nieren für den Organismus ist schwer zu unterschätzen; diese Organe erfüllen nicht nur die Aufgabe, das Blut von Zerfallsprodukten und anderen Stoffen zu reinigen...
Abteilung: Artikel über Gesundheit
Der Sommer dieses Jahres in Russland verlief sehr wechselhaft: Die Regionen waren zunächst unerbittlicher Hitze ausgesetzt, gefolgt von Regenperioden und Hagelstürmen; später kehrte die Hitze zurück, diesmal abwechselnd mit Niederschlägen. Solche heftigen Wetterwechsel belasten viele Menschen...
Abteilung: Artikel über Gesundheit
Erschöpfung, Schlafmangel, Ernährungsfehler, schlechte Stimmung und Wetterlaunen schlagen sich im Äußeren negativ wider. Besonders leidet die Haut: Sie wird rötlich, verliert ihre gesunde Farbe, ist von Milien bedeckt; es erscheinen Wassergeschwülste und dunkle Augenringe. Dass man den Folgen aggressiver Faktoren nicht immer gewachsen ist, liegt auf der Hand; wir können sie jedoch minimieren. Zu diesem Zweck werden kosmetische Mittel und Verfahren eingesetzt, die helfen...
Abteilung: Artikel über Gesundheit
Nächtliche Albträume zählen zu den unangenehmsten Schlafstörungen. Nach Statistiken treten sie bei 4 % der Erwachsenen und fast bei 70 % der Kinder auf...
Abteilung: Artikel über Gesundheit
Die reaktive Pankreatitis ist eine Erkrankung, die durch einen entzündlichen Prozess in der Bauchspeicheldrüse gekennzeichnet ist und meist auf exzessive Aktivität von Verdauungsenzymen zurückzuführen ist. Es handelt sich um einen Sonderzustand, dessen Behandlung chirurgisch erfolgen sollte...
Abteilung: Artikel über Gesundheit
Das menschliche Auge ist täglichen enormen Belastungen ausgesetzt. Die Erhaltung der Sehkraft hängt direkt von einer ausreichenden Versorgung des Augengewebes mit Sauerstoff und Nährstoffen über viele Jahre ab. Diese Aufgabe erfüllen die kleinen Gefäße, die Kapillaren. Für das normale Funktionieren des Sehapparats ist entscheidend, dass sie ihre Integrität bewahren; dies gelingt jedoch keineswegs immer. Mikrotraumen der Augengefäße führen häufig zu kleinen Blutergüssen...
Abteilung: Artikel über Gesundheit
Zelltüchtigkeit – ein weit verbreiteter kosmetischer Mangel, der bei etwa 80 % der Frauen spät auftritt; das Auftreten jeglicher...
Abteilung: Artikel über Gesundheit
Das Kind, das kürzlich geboren wurde, ist von der Liebe seiner Eltern und deren Sorge umgeben; ohne diese Fürsorge kann es nicht überleben. Manche Eltern glauben fälschlicherweise, dass zarte Zuneigung und Liebkosung allein ausreichen, damit sich das Kind richtig entwickelt und glücklich wird...
Abteilung: Artikel über Gesundheit
Die meisten gynäkologischen Erkrankungen zeichnen sich durch drei Hauptmerkmale aus, die jeweils auf einen Besuch beim Gynäkologen hinweisen. Eine genaue Diagnose ist erst nach einer Untersuchung möglich; aufgrund bestimmter Merkmale kann jedoch das Vorhandensein dieser oder jener Pathologie vermutet werden. Im Folgenden betrachten wir die Symptome weiblicher Erkrankungen, die am häufigsten auftreten....
Abteilung: Artikel über Gesundheit
Das Baden mit Suden aus Heilfarben und Pflanzen (Phyto-Bäder) war bereits seit der Zeit Kleopatras verbreitet...
Abteilung: Artikel über Gesundheit
Für viele Ehepaare ist die Frage der Familienplanung von wesentlicher Bedeutung; an erster Stelle steht dabei das Problem der Auswahl wirksamer und sicherer Kontrazeptiva. Russen greifen bis heute selten auf Vasektomie zurück, während diese Methode in den USA außerordentlich populär ist...
Die Abteilung: Artikel über Gesundheit
Es scheint, als hätten Sie den Knirps vor kurzem aus dem Entbindungsheim gebracht; doch die Zeit ist geflogen, und er steht nun am Vorabend des ersten Zusammentreffens mit dem Kinderkollektiv. Wie bereitet man das Kind auf einen Gartenbesuch vor? Was muss gelehrt werden, um den Anpassungsprozess zu erleichtern? Welche Verhaltensweisen sind wichtig, damit der Kleine die Lebensveränderungen schmerzlos übersteht? Wir werden uns bemühen, Antworten auf diese Fragen zu finden.
Die Abteilung: Artikel über Gesundheit
Die Nieren erfüllen die wichtigste Funktion der Blutreinigung von jenen Stoffwechselprodukten, die vom Organismus nicht verwertet werden können.
Die Abteilung: Artikel über Gesundheit
Bewohner großer Städte leiden häufig an einer Erkrankung, die als Syndrom der langdauernden Ermüdung (SCHU) bekannt ist. Dieses Leiden betrifft Menschen aus verschiedenen sozialen und demographischen Gruppen auf allen Kontinenten. Noch mehr Fälle von SCHU werden beobachtet...
Die Abteilung: Artikel über Gesundheit
Das Befinden des Menschen hängt von vielen Faktoren ab. Eine der wichtigsten, jedoch nicht zwingenden, ist die Bewegungsaktivität. Bei Vorhandensein verschiedener Leiden beraten Experten Patienten häufig dazu, sich mit Seefahrt zu beschäftigen, was nachweislich eine hohe Wirksamkeit bei der Genesung aufweist und relativ wenige Gegenanzeigen besitzt. Heute werden wir über die Hauptrichtungen der therapeutischen Wirkung von Seefahrt auf den menschlichen Organismus sprechen.
Die Abteilung: Artikel über Gesundheit
Der Sommer ist in vollem Gange. Viele befinden sich im Begriff, ihren Urlaub im Ausland zu verbringen. Auf die Reisenden warten sanfte Meere, Erholung an den Stränden und die Besichtigung von Sehenswürdigkeiten...
Abteilung: Artikel über Gesundheit
Tatsächlich leiden viele Menschen unter lästigen, ziehenden und stechenden Schmerzen im Rücken, die auf eine Unterkühlung der Muskulatur zurückzuführen sind. In einigen Fällen geht der entzündliche Prozess über das unangenehme Gefühl hinaus und manifestiert sich durch Wassergeschwülste, Indurationen sowie Fieber...
Abteilung: Artikel über Gesundheit
Die Verbesserung des Zustands von Patientinnen nach der Anwendung von Präparaten ohne aktive Wirkstoffe, bekannt als Placebo-Effekt, ist seit langem dokumentiert. Ende des 18. Jahrhunderts begann der amerikanische Arzt Perkins mit der Behandlung von Menschen mittels sogenannter „wunderbarer" Stäbchen aus Stahllegierung und Messing. Es genügte, diesen Gegenstand einige Minuten auf den schmerzenden Punkt zu drücken, um dem Patienten erhebliche Linderung zu verschaffen. Verdächtigt wegen Kurpfuscherei, versuchten seine Kollegen, dieses „Wunder" mit Hilfe der Stäbchen zu reproduzieren...
Abteilung: Artikel über Gesundheit
Rüben, Rettich und Meerrettich – einst genossen diese und andere Lebensmittel bei unseren Vorfahren große Beliebtheit nicht nur als Nahrungsmittel, sondern auch in anderen Bereichen...
Abteilung: Artikel über Gesundheit
Geschäftsfrauen und werdende Mütter stehen vor einer Vielzahl von Herausforderungen. Das wesentliche Dilemma besteht darin, wie das Lieblingskind mit der Arbeit vereinbart werden kann. Welche Fallstricke lauern auf die arbeitende Mutter, wenn sie sich bestimmten Verhaltensanforderungen unterwerfen muss?...
Abteilung: Diashow
Viele Eltern stoßen bei Kindern im Alter von 2 bis 4 Jahren auf übermäßig launisches Verhalten. Das Kind leidet durch ständiges Weinen und Launen nicht nur der Eltern, sondern auch selbst. Worin liegen die Gründe für Kinderlaunen? Wie kann man damit umgehen?...
Abteilung: Slideshow
Der Zustand des menschlichen Rückgrats ist von großer Bedeutung; es dient nicht nur als Stütze für den Körper, sondern enthält auch...
Abteilung: Artikel über Gesundheit
Die Diagnosestellung war und bleibt ein zentrales Problem in der Medizin. Der Behandlungserfolg hängt davon ab, wie genau die Ursache des Unwohlseins bestimmt wird, obwohl die Mehrheit der diagnostischen Methoden bereits genutzt wird...
Abteilung: Artikel über Gesundheit
Musiktherapie ist eine Behandlungsform, deren heilsame Wirkung für den Organismus durch zahlreiche Studien wissenschaftlich bestätigt wurde. Obwohl diese Methode zunächst auf Kontroversen hinsichtlich ihrer Effektivität stieß, ist sie seitdem in vielen Ländern Teil der komplexen Therapie bei kardiovaskulären und respiratorischen Erkrankungen, Rückenschmerzen sowie Wirbelsäulenleiden, psychosomatischen Störungen und weiteren Leiden. Besonders weit verbreitet ist sie in der Pädiatrie.
Abteilung: Artikel über Gesundheit