





Retinoblastom
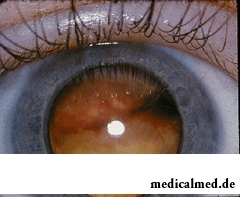
Das Netzhautmalignom – das Retinoblastom besitzt eine neuroektodermale Herkunft und zählt zu den erblichen Erkrankungen. Es wird autosomal-dominant von den Eltern auf die Kinder vererbt.
Die Häufigkeit des Retinoblastoms bei Kindern hat sich in der modernen Ära signifikant erhöht. Zum Vergleich: Während vor zwei Jahrzehnten laut offiziellen medizinischen Statistiken ein Fall pro 30.000 lebendgeborenen Kindern diagnostiziert wurde, liegt die aktuelle Inzidenz bei 1 bis 15 Fällen pro 100.000 Lebendgeborene.
Das Retinoblastom kann sich in jedem Bereich der Netzhaut bilden. Im Anfangsstadium zeigt die Neubildung den reflektierten Lichtglanz des Augenhintergrunds. Im nächsten Stadium entwickelt sich eine flache, trübe Tumormasse mit unscharfen Konturen. Die weitere Entwicklung des Tumors hängt von seiner Histologie und dem allgemeinen Krankheitsbild ab.
Histologie des Retinoblastoms
Experten unterteilen das Retinoblastom je nach Art der Gewebenausbreitung in zwei Stadien:
1. Stadium der Endophytie (intrakulär): Das Malignom wächst hierbei innerhalb des Augapfels. Die Tumorgewächse werden von einer Erhöhung des Augeninnendrucks, dem Auftreten eines Glaukoms und manchmal vom Eintritt der vollen Blindheit begleitet. Der Patient leidet unter Schmerzen, Schwindel und Übelkeit; es entstehen periodisch Brechreizreaktionen, in einigen Fällen kann sich eine anhaltende Anorexie bilden.
2. Stadium der Exophytie (extraokulär): Auf diesem Krankheitsstadium wird das bösartige Wachstum nicht durch den Augapfel begrenzt. Die Tumormassen breiten sich über den Sehnerv ins intrakranielle Gebiet aus; die Metastasen befallen die Lymphknoten.
In einem Auge kann gleichzeitig die Größe beider oben genannten Typen diagnostiziert werden.
Es gibt auch eine Klassifikation des Retinoblastoms nach der Ätiologie der Erkrankung:
1. Erbliches Retinoblastom: Dies ist die häufigste Form der Erkrankung (über 70 % aller Fälle). In der Regel entwickelt sich das Retinoblastom bei Kindern mit weiteren pluralen angeborenen Entwicklungsstörungen – wie Herzfehlern, kortikaler Hyperostose oder einem weichen Gaumen usw. Das maligne Tumorwachstum beim erblichen Retinoblastom zeichnet sich durch eine extrem schnelle Entwicklung aus und betrifft gleichzeitig beide Augen.
Der häufigste Typ des Retinoblastoms tritt im frühen Kindesalter auf (bis zu 30 Monaten).
2. Sporadisches Retinoblastom: Dieser Erkrankungs-Typ ist deutlich seltener und weist eine günstigere Prognose auf. Beim sporadischen Retinoblastom ist nur ein Auge betroffen; die Neubildung entwickelt sich lokal, d.h. es bildet sich eine Tumormasse (einseitige Geschwulst). Das Retinoblastom der sporadischen Form kann im späteren Alter auftreten.
Ursachen des Retinoblastoms
Die Ätiologie des Retinoblastoms ist bis heute nicht vollständig geklärt, insbesondere bei den erblichen Formen. Die alarmierende Zunahme der Erkrankungszahlen deutet auf negative Auswirkungen der modernen ökologischen Situation hin: Eine schlechte Qualität der von der Mehrheit konsumierten Lebensmittel kann offenbar Genmutationen im intrauterinen Stadium der Embryonalentwicklung auslösen.
Symptome des Retinoblastoms
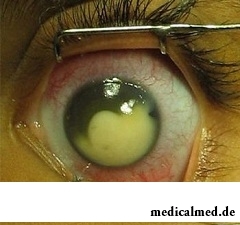
Das Retinoblastom manifestiert sich durch folgende klinische Zeichen:
Rötung des Auges
Schmerzsyndrom
Ausgedehnte Augenhintergrundveränderungen
Teilweise oder vollständige Sehstörung
Gelegentliches Schielen
Leukokorie im Spätstadium – bezeichnet als ‚das weiße Auge‘ (der Pupillenreflex ist nicht rot, sondern weiß).
Diagnostik des Retinoblastoms
Die Diagnostik des Retinoblastoms erfolgt in spezialisierten Zentren. Aufgrund des frühen Alters der Patientinnen werden alle diagnostischen Verfahren und Eingriffe unter Allgemeinanästhesie durchgeführt. In der Regel bevorzugen Ärzte, zytologische und histologische Proben aus dem betroffenen Gewebe nicht zu entnehmen, da während der Manipulation das Risiko einer instrumentellen Verletzung mit hohem metastasierungsgefährdenden Potenzial besteht.
Zur Diagnosestellung des Retinoblastoms sind folgende zwingenden Untersuchungen erforderlich:
Computertomographie (CT) des Gehirns, des Augenhintergrunds und der Orbita;
Ultraschalluntersuchung (Sonographie)
Röntgenaufnahmen
zweiprojektionale Sonographie beider Augen
Magnetresonanztomographie (MRT)
Einsatzmöglichkeiten weiterer diagnostischer Verfahren
Knochenmarkpunktion
Lumbalpunktion
Knochenszintigraphie
Therapie des Retinoblastoms
Die Therapiestrategie beim Retinoblastom orientiert sich an der allgemeinen klinischen Einschätzung des Krankheitszustands sowie am Stadium des Tumors.
Die Experten unterteilen das Retinoblastom in Stadien entsprechend den folgenden Schweregraden:
Stadium I: Der Tumor ist auf die Grenzen des Augenhintergrundes beschränkt.
Stadium II: Der Tumor ist auf den Augapfel beschränkt.
Stadium III: Extraokular.
Stadium IV: Charakterisiert durch Fernmetastasierung.
Nach der Bestimmung des Schweregrades des Retinoblastoms werden folgende Therapiemaßnahmen eingeleitet:
1. Chirurgische Operation: Als wirksame Maßnahme wird sie bei vollständigem Verlust der Sehfunktion und bei Aussichtslosigkeit einer Wiederherstellung eingesetzt. Als komplexe Behandlung werden chemotherapeutische und radiologische Verfahren verordnet.
2. Kryodestruktion – Therapie mittels niedriger Temperaturen beim Retinoblastom des leichten Stadiums im vorderen Bereich der Netzhaut.
3. Fotokoagulation: Die Nutzung von Laserstrahlen erzielt einen guten therapeutischen Effekt bei der Behandlung eines Retinoblastoms des leichten Stadiums im hinteren Bereich der Netzhaut.
4. Thermotherapie (komplexe Anwendung der Mikrowellentherapie sowie Infrarot- und Ultraschallstrahlung).
4. Thermotherapie (komplexe Anwendung der Mikrowellentherapie sowie Infrarot- und Ultraschallstrahlung).

Manchmal scheint es so, als sei die moderne Gesellschaft in zwei Lager geteilt: Die Vertreter des einen Lagers sind überzeugt, dass für eine Kontrazeption etwas Bestimmtes erforderlich ist...
Abteilung: Artikel über Gesundheit
Die Winterseefahrt in offenen Gewässern, im russischen Sprachgebrauch als „Morschewanie" bezeichnet – eine offiziell anerkannte Sportart und eine der extremsten Methoden zur Körperhärtung. Diese Beschäftigung hat eine lange Geschichte und viele Anhänger in vielen Ländern. Jährlich werden...
Abteilung: Artikel über Gesundheit
Eine große Anzahl von Menschen hält an den Prinzipien einer vegetarischen Ernährung fest. Die Frage, wie zu verfahren ist, wenn in der Familie eines Vegetariers Kinder geboren werden und ob es möglich ist, sich ebenso wie die Eltern zu ernähren oder ob der kindliche Organismus nicht für eine außergewöhnlich pflanzliche Nahrung verwendet wird, soll hier geklärt werden.
Abteilung: Artikel zur Gesundheit
Vor 10 bis 15 Jahren galt das Vorhandensein von Computern in russischen Wohnungen als Seltenheit; die Büroräume befanden sich lediglich auf dem Dachboden.
Abteilung: Artikel zur Gesundheit
Der Sommer dieses Jahres in Russland verlief sehr wechselhaft: Die Regionen litten unter der unbarmherzigen Hitze, gefolgt von Regengüssen und Hagelstürmen, woraufhin die Hitze wiederkehrte und sich mit den Niederschlägen abwechselte. Solche heftigen Wetterveränderungen betreffen viele Menschen...
Abteilung: Artikel zur Gesundheit
Es ist seit dem tiefen Altertum bekannt, dass bestimmte Toxine bei oraler Aufnahme in minimalen Mengen therapeutische Wirkungen entfalten. Viele als giftig anerkannte Substanzen werden heute in Heilzwecken eingesetzt und bilden die Hauptkomponenten der von der Pharmaindustrie offiziell vertriebenen Medikamente. Wir berichten hier nur über einige der bekanntesten...
Abteilung: Artikel zur Gesundheit
Die Sklera und die Schleimhaut des Auges sind durchzogen von Blutgefäßen, deren Aufgabe es ist, das Nervengewebe des Organs intensiv zu versorgen...
Abteilung: Artikel zur Gesundheit
Die Praxis der Hypnose wirkt sich auf das menschliche Bewusstsein aus. Über zwei Jahrtausende haben Gelehrte dazu beigetragen, das Phänomen besser zu verstehen und damit Patientinnen unterstützt, die unter schweren Leiden leiden...
Abteilung: Artikel zur Gesundheit
Der Mensch sowie alle anderen Lebewesen auf unserem Planeten nehmen Wetterveränderungen wahr. Es ist normal, dass dies beim gesunden Menschen keine besonderen Beschwerden verursacht. Eine meteorosensitive Reaktion hingegen ist ein pathologischer Zustand, der sich durch eine Verschlimmerung langdauernder Leiden bei Änderungen der Lufttemperatur, des Luftdruckgradienten, der Windstärke sowie magnetischer Stürme und anderer Naturereignisse manifestiert, die die Natur hervorbringt. Personen, die an einer meteorosensiblen Reaktion leiden, sollten...
Abteilung: Artikel zur Gesundheit
Was verstehen wir unter Unkraut? Damit bezeichnet man Pflanzen, die üblicherweise nur für Kompostgruben oder als Futtermittel geeignet sind...
Abteilung: Artikel zur Gesundheit
Ein strahlendes Hollywood-Lächeln kann jeder prahlen. Selbst bei Personen, die regelmäßig den Zahnarzt aufsuchen und auf die Gesundheit ihrer Mundhöhle achten, können sich periodisch Probleme entwickeln: Der Zahnschmelz dunkelt unter dem Einfluss bestimmter Lebensmittel nach, wobei sich Ablagerungen bilden...
Abteilung: Artikel zur Gesundheit
Der Sommer in seiner Hochphase. Viele planen einen Urlaub im Ausland. Die Reisenden erwarten sanfte Meere, Erholung an den Stränden, Besichtigungen von Sehenswürdigkeiten sowie Wanderungen in natürliche und kulturelle Naturschutzgebiete. Leider können jedoch während der Erholungsreise gesundheitliche Probleme auftreten. Im Ausland kann man auf Erkrankungen stoßen, die nicht nur den langgeplännten Urlaub beeinträchtigen, sondern auch eine Behandlung über mehrere Monate nach dem Reiseende zwingen. Eine vollständige Versicherung gegen solche Unannehmlichkeiten ist ratsam...
Abteilung: Artikel zur Gesundheit
Die Geburt ist eines der wichtigsten Ereignisse im Leben jeder Frau. Doch wir Frauen gebären das neue Männlein ins Licht. Zur Zeit...
Abteilung: Artikel über Gesundheit
Energiesparende Lampen gehören zu den populärsten innovativen Technologien, was nicht verwunderlich ist, da sie wirtschaftlicher und langlebiger als herkömmliche Glühlampen sind; gleichzeitig bestehen jedoch Befürchtungen, dass diese Lampen gesundheitsschädigend wirken könnten...
Abteilung: Artikel über Gesundheit
Wir stellen Ihnen einen Überblick über Medikamente vor, die fördernd auf die Potenz wirken, also auf die Fähigkeit des Mannes zur Vollziehung des Geschlechtsakts. Es ist sofort zu sagen, dass nicht immer Verstöße der erektilen Funktion durch die Einnahme eines bestimmten Präparats korrigiert werden können; die Ursachen für eine Senkung der Potenz reichen von banaler Übermüdung bis hin zu einem Tumor im kleinen Becken, weshalb ein Mann bei häufigem Auftreten solcher Probleme einen Arzt konsultieren sollte...
Abteilung: Artikel über Gesundheit
Die Bedeutung der Nieren für den Organismus ist schwer zu unterschätzen; diese Organe erfüllen nicht nur die Aufgabe der Blutreinigung von Abbauprodukten und Ausscheidungsstoffen...
Abteilung: Artikel über Gesundheit
Weiße Zähne und das Hollywood-Lächeln sind der Traum vieler Menschen. Lange Zeit wurde angenommen, dass Zahnverfärbungen und -schäden das Schicksal von Personen sind, die sich falsch ernähren, rauchen und schlecht putzen; jedoch existiert ein Paradoxon: trotz der heutigen Vielfalt an Zahnpasten...
Abteilung: Artikel über Gesundheit
Die Saison der Virusinfektionen ist im Höhepunkt. Jeder kann krank werden, aber die Wahrscheinlichkeit dieses unangenehmen Ereignisses lässt sich minimieren. Es gibt eine Reihe von Maßnahmen, die entweder vollständig helfen oder zumindest die Ansteckung mit Influenza vermeiden bzw. das Leiden in einer leichten Form ohne wesentliche Komplikationen verlaufen lassen; über diese Prophylaxe-Maßnahmen wird im Folgenden gesprochen...
Abteilung: Artikel zur Gesundheit
Das kleine Kind, das kürzlich geboren wurde, ist von der Liebe seiner erwachsenen Familienmitglieder sowie ihren Sorgen umgeben; ohne diese Unterstützung kann das Kind nicht existieren.
Abteilung: Artikel zur Gesundheit
Das Baden in Sud mit Heilfarben und Pflanzen (Phyto-Bäder) war bereits seit den Zeiten Kleopatras verbreitet, die sich auf Schönheit und Gesundheit konzentrierten. Heute ist dies ein einfaches und verfügbares Mittel, das nicht nur nervöse Abmagerungen ermöglicht.
Abteilung: Artikel zur Gesundheit
Moderne Schuhe sind außerordentlich vielfältig. Sie dienen seit langem nicht mehr ausschließlich dem Schutz der Beine. Heute wählen Menschen Stiefel, Sandaletten oder andere Modelle primär aufgrund von Ästhetik, Marke und Stil als Ergänzung zur Kleidung, weniger jedoch wegen Bequemlichkeit und Funktionalität. Beim Kauf wird oft wenig an Sicherheit gedacht; viele populäre Modelle können erhebliche Gesundheitsrisiken verursachen.
Abteilung: Artikel zur Gesundheit
Die Kiefer ist eine der am weitesten verbreiteten Pflanzen unserer Wälder. Ihre Nadeln und das Harz, das nicht umsonst „Schwizer" genannt wird, dienen als wertvolle Heilmittel.
Abteilung: Artikel zur Gesundheit
Zellulitis ist ein sehr häufiger kosmetischer Mangel, der bei Frauen früh oder etwa im Alter von 80 % später auftritt. Sein Auftreten ist mit dem Strukturwandel des subkutanen Fettgewebes verbunden; dabei zeigt sich an der Hautoberfläche zunächst eine Ungleichmäßigkeit (Rundung).
Abteilung: Artikel zur Gesundheit
Es ist unmöglich, sich ein Leben ohne Pflanzen vorzustellen. Tatsächlich finden sich Zimmerpflanzen in fast jeder Wohnung und Produktionsraum; Millionen Menschen widmen sich gärtnerischen Tätigkeiten und dem Gemüseanbau, während viele Stadtbewohner ihre Freizeit auf dem Land verbringen. Dennoch achten wir selten auf jene Eigenschaften unserer grünen Nachwuchspflanzen, die die Umgebung unangenehm oder sogar gefährlich machen können.
Abteilung: Artikel zur Gesundheit
Tee wird von fast allen geschätzt und genutzt. Dieses Getränk verfügt über tonisierende Eigenschaften und enthält Gerbstoffe, die verdauungsfördernd wirken...
Abteilung: Artikel zur Gesundheit
Die Blasenentzündung (Zystitis) ist eine Entzündung der Schleimhaut der Harnblase. Aufgrund bestimmter anatomischer Besonderheiten des weiblichen Urogenitalsystems betrifft diese Erkrankung Frauen etwa viermal häufiger als Männer. Zur Hauptrisikogruppe gehören...
Abteilung: Artikel zur Gesundheit
Die nächste Influenza-Epidemie löst Panik aus; im darauffolgenden Jahr unterliegen wir diesen Manipulationen: der professionell beunruhigten Stimme des Nachrichtensprechers, Zusammenfassungen mit der Zählung verstorbener Patientinnen, Interviews mit Fachpersonal in weißen Kitteln und Werbung für antivirale Mittel unterschiedlicher Wirksamkeit. Dieses Szenario erinnert an Hollywood-Filme über Epidemien, die unseren Planeten bedrohlich zerstören sollen. Zudem besteht eine weitere Ähnlichkeit zum Kino: Alles geht gut zu Ende. Folglich lernen wir uns mit dem Geschehenenden zurechtfinden...
Abteilung: Artikel zur Gesundheit